
Quy trình xây dựng, tối ưu công thức và quy trình sản xuất chế phẩm dược là một trong số những bước quan trọng trong việc nghiên cứu và phát triển sản phẩm dược. Ở bài viết dưới đây, PSB College sẽ chia sẻ một số thông tin chi tiết về các quy trình nói trên – Bài viết được trích từ tài liệu ”Nghiên cứu và phát triển dược phẩm” của Pharma Lab – Dược sĩ Thắng.

1. Xây dựng công thức, quy trình sản xuất và tiêu chuẩn ban đầu
1.1. Xây dựng công thức ban đầu
Viên nén, viên nang và thuốc rắn khác
* Cách 1: Dựa vào các handbook – Xem lại Bộ Handbook về công thức thuốc và quy trình sản xuất (Handbook of pharmaceutical manufacturing formulations – Second edition) trong phần “1.2. Tính giá thành sản phẩm và đánh giá hiệu quả của đề tài”. Xem Volume 1 và 2.
* Cách 2: Cách xây dựng công thức viên nén (viên nén, viên nén bao phim) khi chỉ biết dược chất và danh sách tá dược hoặc khi đã có các nghiên cứu có sẵn trên thế giới. Truy cập vào đường link sau để đọc bài viết:
https://thangtv.net/duoc-va-toi/cach-xay-dung-mot-cong-thuc-vien-nen-co-ban-vien-nen-va-vien-nen bao-phim.html
Thuốc tiêm, dung dịch nhỏ/ xịt mắt, mũi.
* Cách 1: Dựa vào các handbook – Xem lại Bộ Handbook về công thức thuốc và quy trình sản xuất (Handbook of pharmaceutical manufacturing formulations – Second edition) trong phần “1.2. Tính giá thành sản phẩm và đánh giá hiệu quả của đề tài”. Xem Volume 6. Sterile Products * Cách 2: Khi Handbook không có sẵn công thức, các bạn có thể đọc và làm theo bài viết trong đường link sau đây: bài viết về cách xây dựng công thức và quy trình sản xuất của một thuốc tiêm. Truy cập vào đường link sau để đọc bài viết:
https://thangtv.net/duoc-va-toi/thuoc-tiem-lieu-co-kho-cong-thuc-va-quy-trinh-san-xuat-thuoc tiem.html
Thuốc lỏng đường uống: dung dịch, hỗn dịch, nhũ tương, siro…
Thuốc bán rắn: cream, nhũ tương…
Đối với các dạng bào chế thuốc lỏng đường uống và thuốc bán rắn, các bạn tiến hành tương tự như với viên nén, viên nang và thuốc tiêm. Tham khảo Handbook volume 3 và 4, truy cập vào các nghiên cứu nước ngoài có sắn và cuối cùng là tự đưa ra công thức dựa vào kinh nghiệm và lý thuyết. (Phần này sẽ được cập nhật trong những Phiên bản sau của Tài liệu này).
Email: pharmalabs.rd@gmail.com Pharma Labs: FB.com and Youtube
Tài liệu: Nghiên Cứu Và Phát Triển Dược Phẩm 9 / 18 Dược sĩ Thắng: https://thangtv.net
1.2. Xây dựng quy trình sản xuất ban đầu
1.2.1. Viên nén, viên nang và thuốc rắn khác
Có 3 quy trình sản xuất phổ biến bao gồm:
* Dập thẳng (đóng nang trực tiếp):
Kỹ thuật dập thẳng là kỹ thuật đơn giản nhất, quy trình sản xuất chỉ bao gồm những giai đoạn sau: Ngoài ra có thể có thêm các loại tá dược khác như tá dược dính, tá dược tăng độ tan …. Chi tiết kỹ thuật dập thẳng cho viên nén và đóng nang trực tiếp, các bạn xem trong bài viết sau: https://thangtv.net/duoc-va-toi/dap-thang-trong-san-xuat-vien-nen-mo-ta-chi-tiet-qtsx-va-truong-hop ap-dung.html
1.2.2. Làm sao để biết một thuốc sẽ được dập thẳng, thay vì tạo hạt khô, ướt?
Có 3 loại công thức mà các nghiên cứu viên hay dập thẳng.
Thứ nhất là công thức mà dược chất có tính chất trơn chảy tốt và chịu nén, nghiên cứu viên hoàn toàn có thể dập thẳng hoặc trộn thẳng để đóng nang, giúp tiết kiệm chi phí mà tránh ảnh hưởng tới độ ổn định của dược chất.
Thứ hai là quan sát trong thành phần viên nén, các tá dược dùng chủ yếu là tá dược dập thẳng. Ví dụ: Công thức dùng lactose khan chứ không dùng lactose monohydrate, dùng mannitol DC chứ không dùng mannitol thường …
Thứ ba là tỷ lệ dược chất/tá dược ở mức thấp, từ đó tính chất trơn chảy hay chịu nén sẽ phụ thuộc chủ yếu vào tá dược độn mặc dù dược chất có thể trơn chảy kém và chịu nén kém. Không có một con số chính xác về tỷ lệ dược chất/ tá dược ở mức thấp bao nhiêu thì sẽ làm công thức dập thẳng, điều này cần phải khảo sát/ nghiên cứu hoặc dựa vào kinh nghiệm. Nhưng nếu 1 viên nén 300mg mà dược chất chỉ 50 mg thì khoảng 80% sẽ dập thẳng.
Tuy nhiên, theo ưu tiên nghiên cứu thì nên bắt đầu khảo sát công thức theo phương pháp dập thẳng, nếu dập thẳng không thể tạo ra viên nén đạt yêu cầu thì chuyển sang phương pháp tạo hạt khô, rồi cuối cùng mới là tạo hạt ướt. Vì tạo hạt khô sẽ ảnh hưởng ít hơn tới độ ổn định của dược chất so với tạo hạt ướt.
Email: pharmalabs.rd@gmail.com Pharma Labs: FB.com and Youtube
Tài liệu: Nghiên Cứu Và Phát Triển Dược Phẩm 10 / 18 Dược sĩ Thắng: https://thangtv.net
Tạo hạt khô
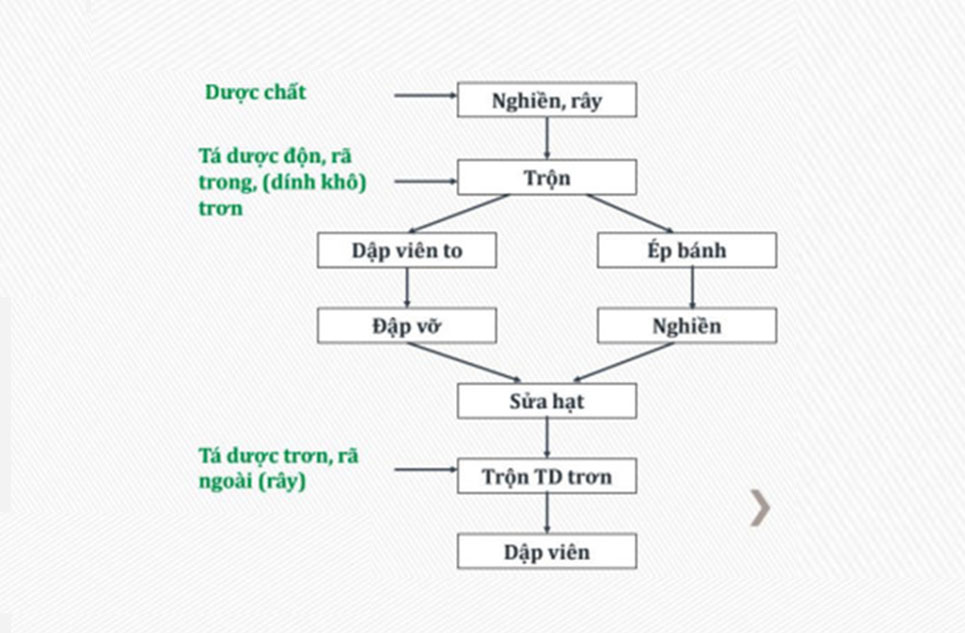
Khác với phương pháp dập thẳng, phương pháp tạo hạt khô sẽ có thêm bước tạo các hạt chứa dược chất. Việc tạo hạt giúp tăng sự trơn chảy cho khối bột, tăng khả năng chịu nén và nhiều vai trò khác nữa.
Có 2 cách tạo hạt khô như trên hình bên trên, cách 1 là dập viên to sau đó đập vỡ. Cách 2 là ép bánh sau đó nghiền. Theo như quan sát tại Việt Nam, phương pháp dập viên to rồi đập vỡ chủ yếu áp dụng ở quy mô thử nghiệm (labs) vì phương pháp áp dụng tốt với lượng mẫu nhỏ. Cách 2 là cán (ép bánh) sau đó nghiền được áp dụng nhiều trên quy mô pilot và công nghiệp, vì trên quy mô công nghiệp có máy cán tạo hạt khô, cũng như việc vận hành máy cán tạo hạt sẽ dễ dàng và tạo ra hạt có kích thước đều hơn.
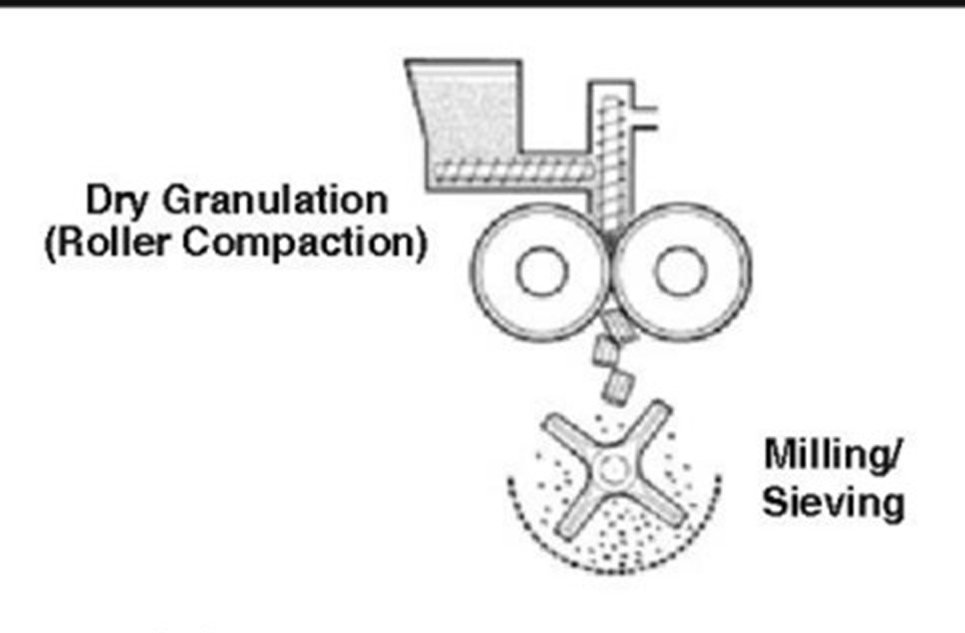
Nguyên lý: Khối bột chứa dược chất sẽ được các trục vít nén sơ bộ và đẩy vào vị trí cán. “ trục cán hình tròn sẽ quay ngược chiều nhau, tác động lên nhau một lực nén và biến khối bột thành các dải,
Email: pharmalabs.rd@gmail.com Pharma Labs: FB.com and Youtube
Tài liệu: Nghiên Cứu Và Phát Triển Dược Phẩm 11 / 18 Dược sĩ Thắng: https://thangtv.net
thanh bột (giống nguyên lý tạo ra viên nén). Thanh bột sau đó sẽ được chuyển qua máy nghiền và mắt rây. Những hạt có kích thước phù hợp sẽ qua được rây => thu được cốm sau khi cán.
Tạo hạt ướt
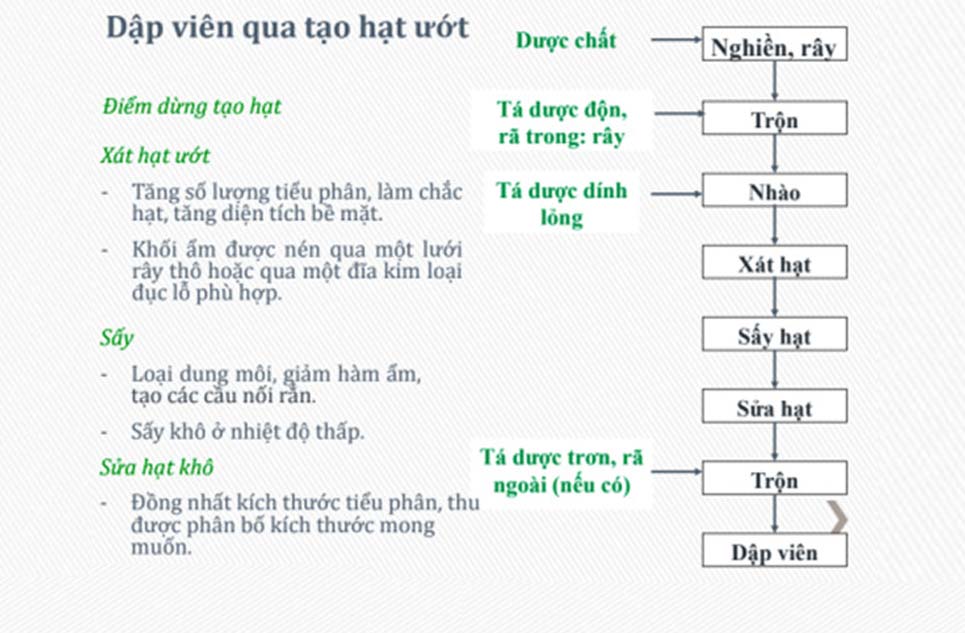
Tạo hạt ướt là phương pháp cuối cùng áp dụng nếu như 2 phương pháp sản xuất dập thẳng và tạo hạt khô không hiệu quả. Vì dược chất sẽ bị tiếp xúc với nhiệt và ẩm.
Lưu ý: Lượng nước cần nhào là bao nhiêu, thời gian nhào, tốc độ nhào, nhiệt độ sấy, thời gian sấy là bao nhiêu thì đều cần phải khảo sát cụ thể.
1.2.3. Thuốc tiêm, dung dịch nhỏ/ xịt mắt, mũi
Quy trình sản xuất thuốc tiêm, thuốc dùng đường mắt, các bạn có thể đọc bài viết này của Pharma Labs để có một cái nhìn tổng quát và hiểu được nguyên lý.
https://thangtv.net/duoc-va-toi/thuoc-tiem-lieu-co-kho-cong-thuc-va-quy-trinh-san-xuat-thuoc tiem.html
1.2.4. Thuốc lỏng đường uống: dung dịch, hỗn dịch, nhũ tương, siro…
1.2.5. Thuốc bán rắn: cream, nhũ tương…
Đối với các dạng bào chế thuốc lỏng đường uống và thuốc bán rắn, sẽ được cập nhật trong phiên bản tiếp theo của tài liệu này.
1.3. Xây dựng tiêu chuẩn ban đầu
– Một tiêu chuẩn sẽ bao gồm: Giới hạn các chỉ tiêu (Specifications) và Phương pháp kiểm nghiệm (Analytical procedure).
1.3.1. Tiêu chuẩn theo dược điển
– Các dược điển hay dùng ở Việt Nam bao gồm: Dược Điển Việt Nam (VP – DĐVN), Dược Điển Anh (BP), Dược Điển Mỹ (USP-NF: USP là dược chất và thành phẩm; NF là tá dược), Dược Điển Châu Âu (EP). Ngoài ra còn một số dược điển ít dụng hơn như: Nhật (JP), Ấn độ (IP), Quốc tế (Int. Phr.), Pháp (FP), Trung Quốc (CP) ….
Email: pharmalabs.rd@gmail.com Pharma Labs: FB.com and Youtube
Tài liệu: Nghiên Cứu Và Phát Triển Dược Phẩm 12 / 18 Dược sĩ Thắng: https://thangtv.net
Để làm sao biết một dược chất, thành phẩm có trong Dược điển nào, các bạn hãy tham khảo bài viết sau nhé:
https://thangtv.net/duoc-va-toi/cach-kiem-tra-duoc-chat-va-ta-duoc-co-trong-duoc-dien-nao pharmacopoeia.html
– Một Dược điển sẽ bao gồm rất nhiều phần, nhưng với giai đoạn đầu này thì chỉ cần quan tâm tới một số phần chính sau:
- Phần chuyên luận chung: như yêu cầu với viên nén, viên nang, thuốc tiêm, thuốc siro …. Phần chuyên luận thành phẩm: như Viên nén paracetamol, dung dịch tiêm paracetamol … Phần chuyên luận dược chất: Như Cefadroxil monohydrate, Cefaclor ….
- Phần phương pháp test chung: Phương pháp đo quang, HPLC …
– Việc xây dựng tiêu chuẩn ban đầu sẽ là sự kết hợp của yêu cầu trong chuyên luận chung của dạng bào chế và yêu cầu trong chuyên luận thành phẩm của dược chất đó.
VD:
Chuyên luận viên nén chung yêu cầu của DĐVN:
- Tính chất
- Đồng đều khối lượng hoặc đồng đều hàm lượng
- Độ hòa tan: Yêu cầu trong chuyên luận riêng
- Định lượng: Yêu cầu trong chuyên luận riêng
Chuyên luận riêng của Viên nén Paracetamol trong DDDVN:
- Tính chất
- Định tính
- Độ hòa tan
- 4-Aminophenol
- Tạp chất liên quan
- Định lượng
Như vậy tiêu chuẩn của viên nén Paracetamol sẽ là:
- Tính chất
- Định tính
- Đồng đều khối lượng
- Độ hòa tan
- 4-Aminophenol
- Tạp chất liên quan
- Định lượng
1.3.2. Tiêu chuẩn không theo dược điển
Những thuốc không theo dược điển thì thường là những thuốc còn mới, chưa hết hoặc mới hết hạn bản quyền. Tiêu chuẩn đối với thành phẩm và/ hoặc dược chất chưa được công bố trong các dược điển. Với những thành phẩm không có trong dược điển, tiêu chuẩn xây dựng cho dược chất sẽ dựa trên yêu cầu của chuyên luận chung của dạng bào chế trong Dược điển, yêu cầu trong chuyên luận dược chất (theo dược điển hoặc tiêu chuẩn của nhà sản xuất) và kết hợp với khảo sát các mẫu chứng (thuốc gốc).
Ngoài ra, cũng có thể tìm hiểu các nghiên cứu về phương pháp kiểm nghiệm của các thành phẩm thuốc này trên các tạp chí uy tín, các nguồn tham khảo về nghiên cứu khoa học khác. Theo như kinh nghiệm của Pharma Labs, với những thuốc này thì tiêu chuẩn ban đầu cần xây dựng đạt yêu cầu chung đối với dạng bào chế, thêm các chỉ tiêu mà dược chất có như Tạp chất liên quan, … Các thông số của phương pháp kiểm nghiệm (sử dụng HPLC hay đo quang) thì sẽ dựa chủ yếu vào phương pháp của Dược chất.
Email: pharmalabs.rd@gmail.com Pharma Labs: FB.com and Youtube
Tài liệu: Nghiên Cứu Và Phát Triển Dược Phẩm 13 / 18 Dược sĩ Thắng: https://thangtv.net
VD: Viên nén bao phim A 500mg, Dược chất A cần phải đạt chỉ tiêu tạp chất X < 0.1%, tổng tạp < 0.3%.
Thì theo Dược điển Anh (BP)
Tiêu chuẩn của thuốc A tạm thời là:
- Hình thức
- Định tính
- Đồng đều khối lượng
- Độ hòa tan
- Tạp chất liên quan
- Định lượng
2. Nghiên cứu và tối ưu hóa công thức
2.1. Nghiên cứu tương kỵ dược chất, tá dược
Giai đoạn đầu tiên của nghiên cứu công thức luôn liên quan tới nghiên cứu tương kỵ Dược chất với tá dược để lựa chọn tá dược có thể dùng trong công thức hay không. Dược chất và tá dược được trộn theo tỷ lệ xác định, sẽ được tiếp xúc với môi trường 4°C và 40°C/ độ ẩm 75% trong vòng 4 tuần. Đựng hỗn hợp trong lọ thủy tinh với nắp LDPE đã được chọc thủng, giúp hỗn hợp tiếp xác với điều kiện bảo quản. Quan sát các hỗn hợp trong 4 tuần để xem có sự thay đổi nào không, so sánh với mẫu chứng được bảo quản ở điều kiện.
Đó là cách cơ bản nhất để nghiên cứu tương kỵ dược chất và tá dược. Với các dạng bào chế khác nhau thì có phương pháp thử nghiệm khác nhau (hòa tan, phân tán …), nhưng nguyên lý chung vẫn là cho tiếp xúc, để trong điều kiện lão hóa để quan sát sự thay đổi so với mẫu chứng (chỉ có dược chất).
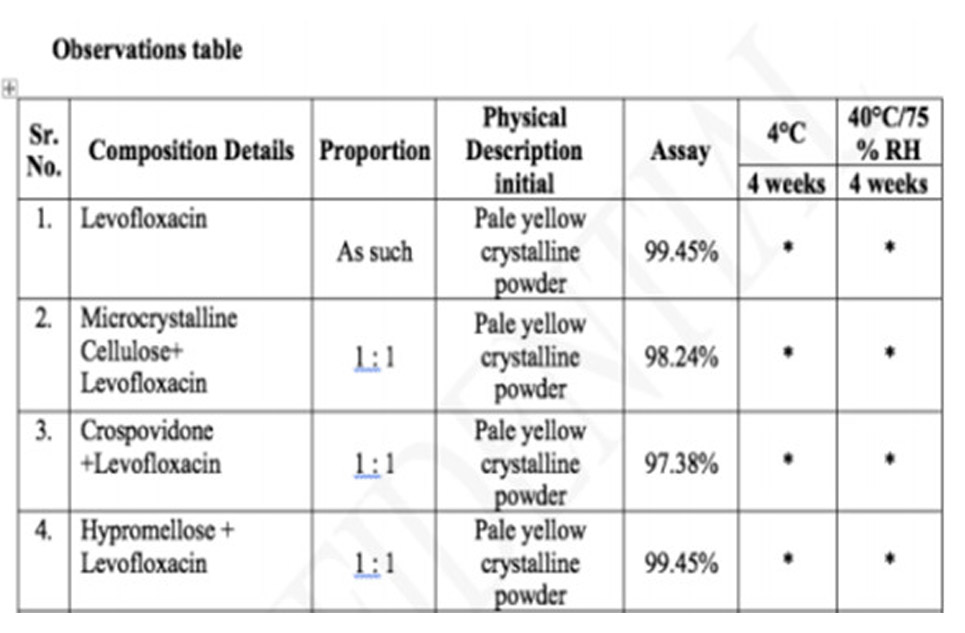
Chỉ thử với những tá dược mình dự kiến tiến hành thử nghiệm trong các công thức sơ bộ đã xây dựng từ bước trước. Nếu tá dược nào bị tương kỵ thì không dùng. Tương kỵ là thế nào: Khối bột bị biến màu, bị ẩm quá, mùi khó chịu hơn: thấy mẫu bị hỏng so với mẫu chứng ở cùng điều kiện.
Email: pharmalabs.rd@gmail.com Pharma Labs: FB.com and Youtube
Tài liệu: Nghiên Cứu Và Phát Triển Dược Phẩm 14 / 18 Dược sĩ Thắng: https://thangtv.net
2.2. Khảo sát các công thức ban đầu
Việc khảo sát các công thức, quy trình sản xuất ban đầu luôn ưu theo thứ tự:
- Công thức ít thành phần, đơn giản nhất. Nếu kiểm nghiệm chỉ tiêu vật lý và/ hoặc hóa học không đạt thì mới thay đổi công thức hoặc thêm các thành phần khác để cải thiện. VD: Công thức viên nén ban đầu có thể không cần đến tá dược rã, nếu viên vẫn rã được thì oke, nếu viên không rã đạt được tiêu chuẩn thì mới phải thêm tá dược rã.
- Quy trình sản xuất đơn giản nhất trước: Ví dụ viên nén sản xuất bằng phương pháp dập thẳng trước, nếu không được thì có thể tạo hạt khô hoặc tạo hạt ướt ….
- Tiêu chuẩn đánh giá: Đánh giá những tiêu chuẩn vật lý, dễ làm trước. Nếu đạt các chỉ tiêu vật lý này thì mới tiến hành với các chỉ tiêu hóa học: Cách này sẽ tiết kiệm thời gian và chi phí nghiên cứu. Ví dụ viên nén thì sẽ thử các chỉ tiêu như Độ cứng, độ mài mòn, độ rã, độ đồng đều khối lượng trước. Khi các chỉ tiêu đó đã đạt thì mới tiến hành đánh giá hàm lượng, độ hòa tan. Hàm lượng, độ hòa tan mà đạt thì mới đánh giá tạp chất liên quan.
Việc nghiên cứu, khảo sát các công thức ban đầu sẽ giúp đưa ra được một công thức sơ bộ với các thành phần và lượng của các thành phần ấy. Khi sản xuất theo quy trình đã khảo sát thì đạt các chỉ tiêu ban đầu đề ra. Công thức này sẽ được đưa ra làm đầu vào cho giai đoạn tối ưu hóa công thức. Note: Công thức của giai đoạn này cũng có thể mang đi nghiên cứu độ ổn định, sau đó đăng ký luôn mà không cần đi qua giai đoạn tối ưu hóa công thức cũng được, chỉ cần thuốc đạt độ ổn định theo tiêu chuẩn đã được thẩm định.
2.3. Tối ưu hóa công thức
Tối ưu hoá một công thức hay quy trình bào chế là việc tìm công thức, thông số (hay điều kiện tiến hành) của quy trình để sản phẩm làm ra đạt chất lượng tốt nhất trong giới hạn mong muốn của người làm thí nghiệm.
Việc làm tối ưu hóa công thức khá phức tạp, cần sự am hiểu và cần làm các phân tích thống kê. Các bạn có thể tham khảo tài liệu “Một số phương pháp thiết kế thí nghiệm và tối ưu hóa ứng dụng trong bào chế” của Giảng viên Nguyễn Trần Linh – Đại Học Dược Hà Nội:
https://drive.google.com/file/d/13iNYC8wJp8KVOVpyR6dI0Q4-sEpC-Oe3/view?usp=sharing Đọc thêm trong design Space ICH Q8 Pharmaceutical development.
2.4. Sản xuất trên quy mô pilot
Theo Phụ lục I thông tư 32/2018/TT-BYT, quy mô Pilot là các lô được sử dụng trong quá trình phát triển và tối ưu hoá quy trình sản xuất. Quy mô của lô pilot nên tối thiểu là 10% quy mô của lô sản xuất thực tế. Với các dạng thuốc rắn dùng đường uống, cỡ lô pilot cần phải tối thiểu là 10% của lô sản xuất thực tế hoặc 100.000 đơn vị phân liều (giá trị nào lớn hơn sẽ được lựa chọn nếu không có cách lý giải nào khác).
Với các dạng báo chế khác nhau và dược chất khác nhau thì sẽ phải cần số lượng các lô pilot khác nhau. Tham khảo Phụ lục I thông tư 32/2018/TT-BYT để biết thêm chi tiết. Thông thường, cần tối thiểu 2 lô pilot để thẩm định quy trình sản xuất và nghiên cứu độ ổn định thì mới có thể nộp được hồ sơ đăng ký.
Thử nghiệm trên quy mô pilot giúp dự đoán sản phẩm sau này được sản xuất trên quy mô sản xuất có đạt yêu cầu chất lượng và độ ổn định hay không.
Việc sản xuất trên quy mô pilot sẽ tiến hành tương tự như trên quy mô sản xuất, chỉ là ở cỡ lô nhỏ hơn, nên các bạn tham khảo thêm mục 3. Sản xuất và bán hàng để biết thêm cách tiến hành chi tiết.
Email: pharmalabs.rd@gmail.com Pharma Labs: FB.com and Youtube
Tài liệu: Nghiên Cứu Và Phát Triển Dược Phẩm 15 / 18 Dược sĩ Thắng: https://thangtv.net
3. Xây dựng tiêu chuẩn và thẩm định tiêu chuẩn
– Từ tiêu chuẩn ban đầu và kết quả kiểm nghiệm các mẫu thử nghiệm. Tiến hành chạy thử và điều chỉnh tiêu chuẩn cho đến khi thu được kết quả chính xác và lặp lại.
– Tiến hành thẩm định sẽ có sự khác nhau giữa tiêu chuẩn có trong dược điển và tiêu chuẩn không có trong dược điển:
- Với tiêu chuẩn theo dược điển: Cần đánh giá sự phù hợp của hệ thống (system suitability) với các chỉ tiêu cần thẩm định như HPLC, IR, UV-VIS ….
- Với tiêu chuẩn không theo dược điển: Cần đánh giá Sự phù hợp của hệ thống, tính đặc hiệu (specificity), tính chính xác (accuracy), tính lặp lại (repeatability) của phương pháp cần thẩm định.
Một bộ tiêu chuẩn P.5 sẽ bao gồm các thành phần sau:
- Tiêu chuẩn (specifications): P.5.1
- Phương pháp tiến hành (analytical method): P.5.2
- Thẩm định phương pháp phân tích (validation): Bao gồm Đề cương (protocol) + Báo cáo (report) + Dữ liệu thô (raw data: như sắc ký đồ, kết quả…). P.5.3
- Phiếu kiểm nghiệm (Batch analysis) P.5.4
- Phần thuyết minh tiêu chuẩn P.5.5 và P.5.6
4. Nghiên cứu độ ổn định
Các tài liệu có thể tham khảo để hiểu và biết các tiến hành nghiên cứu độ ổn định: – Phụ lục I thông tư 32/2018/TT-BYT
– Hướng dẫn Asean: Asean Guideline On Stability Study Of Drug Product
– ICH:
- Guideline On Stability Testing: Stability Testing Of Existing Active Substances And Related Finished Products
- ICH Topic Q1A (R2): Stability Testing of new Drug Substances and Products. ● ICH Topic Q1C: Stability Testing: Requirements for New Dosage Forms. ● ICH Topic Q1D: Bracketing and Matrixing designs for Stability Testing of Drug Substances and Drug Products.
- ICH Topic Q1E: Evaluation of Stability Data.
- ICH Topic Q1F: Stability Data Package for Registration Applications in Climatic Zones III and IV.
Thông thường, việc nghiên cứu độ ổn định nên được tiến hành từ quy mô nhỏ trước, quy mô vài nghìn viên, gói – tạm gọi là quy mô Lab… để dự đoán liệu công thức có đảm bảo độ ổn định hay không. Sau một khoảng thời gian nghiên cứu trên quy mô Lab (1 tháng đến 6 tháng ở điều kiện cấp tốc), nếu thuốc đạt tiêu chuẩn thì sẽ tiến hành sản xuất trên quy mô pilot và đánh giá độ ổn định trên quy mô này.
Việc đánh giá trên quy mô Lab trước giúp đảm bảo độ ổn định trên quy mô Pilot được thành công, nếu không thành công trên quy mô pilot thì chi phí bỏ ra cho nghiên cứu lần tiếp theo là khá nhiều (một lô pilot của viên nén tối thiểu là 100.000 viên, việc thất bại với lô 100.000 viên là rất tốn kém). Còn tại sao nhất quyết phải nghiên cứu độ ổn định trên quy mô pilot, vì độ ổn định nộp trong hồ sơ đăng ký chỉ chấp nhận từ cỡ lô pilot trở lên, một vài trường hợp thì có thể chấp nhận 2 lô pilot + 1 lô Lab.
Nghiên cứu độ ổn định sẽ tiến hành ở điều kiện cấp tốc và dài hạn (30oC, RH 75% và 40oC, RH 75%) nếu thuốc xuất khẩu sang EU thì cần thêm điều kiện 25oC, RH 60%. Một số thuốc phải bản quản lạnh thì sẽ phải nghiên cứu ở điều kiện khác hơn (ví dụ 2 – 8oC).
Email: pharmalabs.rd@gmail.com Pharma Labs: FB.com and Youtube
Tài liệu: Nghiên Cứu Và Phát Triển Dược Phẩm 16 / 18 Dược sĩ Thắng: https://thangtv.net
Việc nghiên cứu sẽ tiến hành trên các tủ vi khí hậu: Loại tủ kiểm sát nhiệt độ và độ ẩm trong tủ hoặc chỉ kiểm soát nhiệt độ (chỉ kiểm soát nhiệt độ áp dụng cho thuốc có bao bì không thấm như lọ thủy tinh…). Các bạn có thể tìm hiểu về tủ vi khí hậu theo tên tiếng anh là Stability Chamber. Sơ đồ quy trình của nghiên cứu độ ổn định:
Chuẩn bị đề cương nghiên cứu độ ổn định
Stability protocol preparation
Lưu kết quả và tạo báo cáo độ ổn định
Stability record filling and stability report generation
Kết thúc
nghiên cứu độ ổn định Completion of stability study
Kế hoạch lấy mẫu và thông báo lấy mẫu
Sampling plan and sampling notification
Kiểm nghiệm và đánh giá kết quả mẫu độ ổn định Stability sample testing and result evaluation
Nhận mẫu, phân chia và lưu mẫu độ ổn định
stability sample receiving, allocation and storage
Kế hoạch lấy mẫu từ môi trường bảo quản
Sampling plan from stability conditions
Tùy vào sản phẩm thì có độ ổn định 6 tháng ở cấp tốc và 6 tháng điều kiện thực hoặc 6 tháng cấp tốc + 12 tháng điều kiện thực thì đủ dữ liệu để nộp hồ sơ (quy định này có trong phụ lục I thông tư 32 và ICH). Tuổi thọ dự kiến cho thuốc sẽ có thể gấp đôi thời gian nghiên cứu dài hạn nhưng không quá 12 tháng. VD: Nghiên cứu độ ổn định dài hạn được 6 tháng thì hạn dùng đăng ký là 12 tháng, độ ổn định 12 tháng thì hạn dùng là 24 tháng, độ ổn định là 24 tháng thì hạn dùng chỉ là 36 tháng …
Nghiên cứu độ ổn định sẽ gồm: Đề cương, bảng kế hoạch lấy mẫu, báo cáo nghiên cứu độ ổn định và bảng dữ liệu kết quả nghiên cứu độ ổn định.
Xem thêm các bài viết khác:
[Review] Rotuven 300 có thực sự tốt? Công dụng? Giá bán?
[REVIEW] Khang Dược Sâm có tốt không? Công dụng, Giá bán